Proper analysis of complex lasso proteins and (bio)polymers requires investigation of its dynamics. This, on the other hand requires the ability to study in detail also a selected frame. LinkProt provides both options to the users.
The LinkProt server supports a couple of basic formats of input files and gives the user a set of advanced options that allow adjusting this tool to the user's needs. Additionally, the files enabling visualization of the structure with the surface spanned on the closed loops can be downloaded. Currently LinkProt provides files for vmd and Mathematica. Such presentation would certainly help the users to imagine the notion and location of minimal surface spanned on the closed loops and can be essential in further personal analysis.
To further facilitate the understanding of the link-type entanglement, smooth representation of link proteins or (bio)polymer (in pdb format) is available as well. All the details are described in the following subsections:
Input
Coordinates of the structure can be provided in following ways:
1 -9.102 -18.555 15.000 2 -9.384 -17.556 14.080 3 -5.661 -16.841 14.367 4 -3.660 -15.387 11.487 5 0.096 -15.769 11.241 6 2.646 -13.739 9.329 7 5.806 -15.775 8.604 ...
Remark: In pdb file all information should be located in their proper position in line. In particular, the "CA" atom name should be placed in 14th and 15th characters of line. This notation (although not required exactly by pdb standard) is widely used. Please note however, that some format converting software shift the "CA" characters. E.g. CatDCD locates "CA" in 15th and 16th character of the line. Please note also, that the chain name is required to be present in the pdb file.
After selecting the file, the user is supposed to choose the covalent loops, which ought to be analyzed. The indices must be the same as in the file provided. We do not use the atoms renumbering, and accept e.g. negative indices. Always two loops must be specified.
The uploaded file is checked to confirm that it contains proper protein data. The conditions which protein chain must fulfill are basically the same, as in the case of the data stored in the database (see Closed loop detection). Those are:
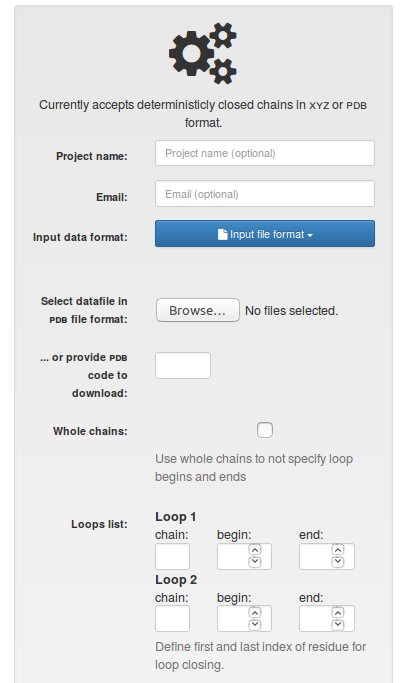
Fig.1 The view of the input page for single structure with "PDB file format" option selected.
Output
The results of the single structure analysis are presented in the same way as the information about any chain in the LinkProt database, described in details in the Single protein chain data presentation section (Fig. 2).
In particular, the user can download all information about a link-type entanglement and visualization facilitating interpretation of the data. In particular users can download the structure in pdb and xyz format, files for vmd, Mathematica and JSmol, enabling them to view the minimal surface spanned on the closed loop. The file for vmd is written as a tcl script. To view the surface, one has to first open the structure in vmd (the downloaded pdb file), click Extensions-> TK Console and load the tcl script in the new window via File->Load file.
Additionally, user can download all files with smooth representation of the structure. This could be helpful in understanding link-type entanglement in more complicated proteins. LinkProt uses the procedure where chain is being smoothed via averaging coordinates of three neighbouring atoms, as long as the link type of the structure stays unchanged (the running average), or 15 runs are done (the sufficient number in all known cases). The number occuring at the end of the “smooth files” names indicates the number of iterations of that procedure that were applied to a chain.
During the analysis there some errors concerning the input file may occure. We provide a full list of known problems with possible solutions in the Possible errors subsection.
A structure uploaded and analyzed by the user is stored for 14 days (so that it can be viewed again).
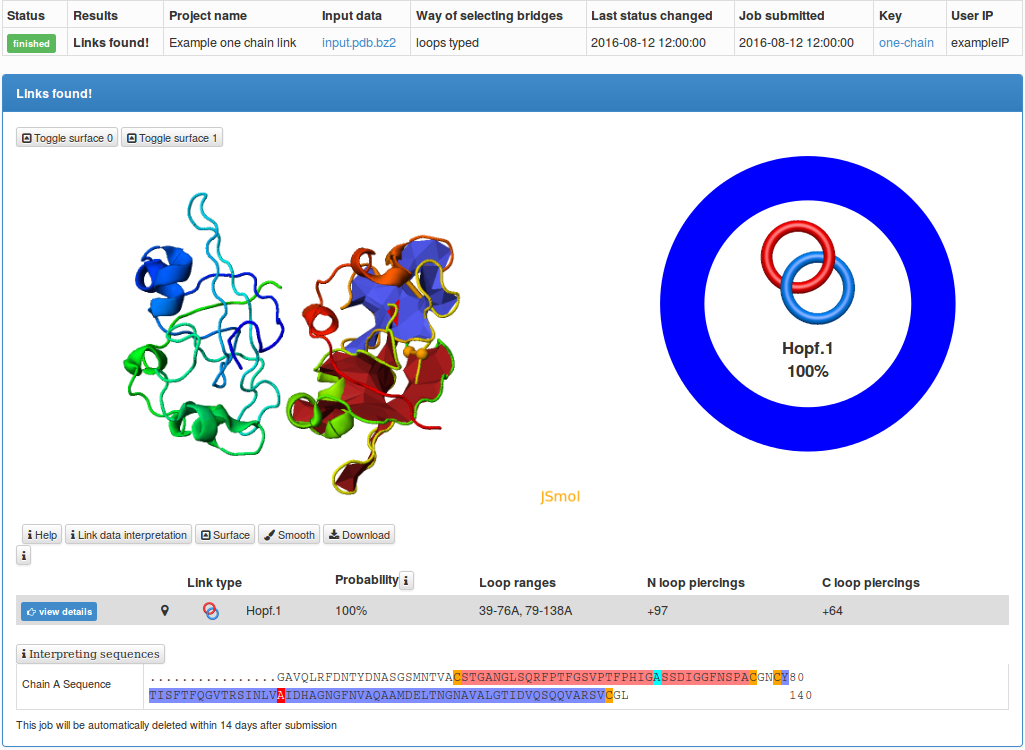
Fig. 2 The typical output for a single structure analysis.
During the analysis some errors concerning input file may occure. We provide a full list of known problems. Clicking on them will display possible solutions. If after reading all known solutions you are sure, that the file is correct and contains the Cα coordinates written in proper way, please send us the file, as that could be a clue how to fix and improve our server.